
The recent findings of DNA fragments in the Pfizer and Moderna Covid-19 vaccines has led many to question why the FDA, which is responsible for monitoring the quality and safety of the vaccines, has failed to sound the alarm.
For years, the FDA has known about the risk posed by residual DNA in vaccines. Its own guidance to industry states:
“Residual DNA might be a risk to your final product because of oncogenic and/or infectivity potential. There are several potential mechanisms by which residual DNA could be oncogenic, including the integration and expression of encoded oncogenes or insertional mutagenesis following DNA integration.”
Put simply, the FDA acknowledges the possibility that fragments of DNA left over by the manufacturing process can be incorporated into a patient’s own DNA, to potentially cause cancer.
FDA and WHO guidelines consider the amount of residual DNA in a single dose of traditional vaccine should not exceed 10 ng (one billionth of a gram).
But this limit – used for traditional vaccines – is unlikely to be relevant to the mRNA vaccines whose lipid nanoparticles can penetrate inside cells to deliver the mRNA efficiently.
A recent preprint paper by Speicher et al analysed batches of the monovalent and bivalent mRNA vaccines in Canada.
The authors found “the presence of billions to hundreds of billions of DNA molecules per dose in these vaccines. Using fluorometry all vaccines exceed the guidelines for residual DNA set by FDA and WHO of 10 ng/dose.”
Speicher et al also reported finding fragments of DNA larger than 200 base pairs (a measure of the length of the DNA) which also exceeds FDA guidelines.
Notably, the authors commented that for the Pfizer product, the higher the level of DNA fragments found in the vaccine, the higher the rate of serious adverse events.
Some experts say the risk of genome integration in humans is very low, but a recent publication in Nature found that around 7 percent of cells are integrated when mixed with a transfection solution containing linear pieces of DNA.
Is the FDA Concerned?
The US Food and Drug Administration (FDA) continues to insist that any residual DNA contamination in the Covid vaccines is not a problem and that it “stands behind its findings of quality, safety, and efficacy for the mRNA vaccines.”
“While concerns have been raised previously as theoretical issues, available scientific evidence supports the conclusion that the minute amounts of residual DNA do not cause cancer, or changes to a person’s genetic code,” added the FDA.
The FDA would not provide the “available scientific evidence” to support its claim, but it’s worth noting that the vaccines’ own product labels show that genotoxicity and carcinogenicity tests were not carried out prior to their use.
David Wiseman, a research bioscientist involved in medical product development and co-author on the study by Speicher et al said the FDA’s claim that there is no evidence of a cancer link is becoming “untenable.”
“The CDC’s own analysis on the vaccine’s safety signal in VAERS shows there could be a signal for some cancers,” said Wiseman pointing to a report he co-authored and sent to the National Academies.
In the table (highlighted in yellow), a safety signal is considered to be significant, and worthy of further investigation, if the value in the column marked PRR exceeds 2 and the value in the Chi-Square column exceeds 4.
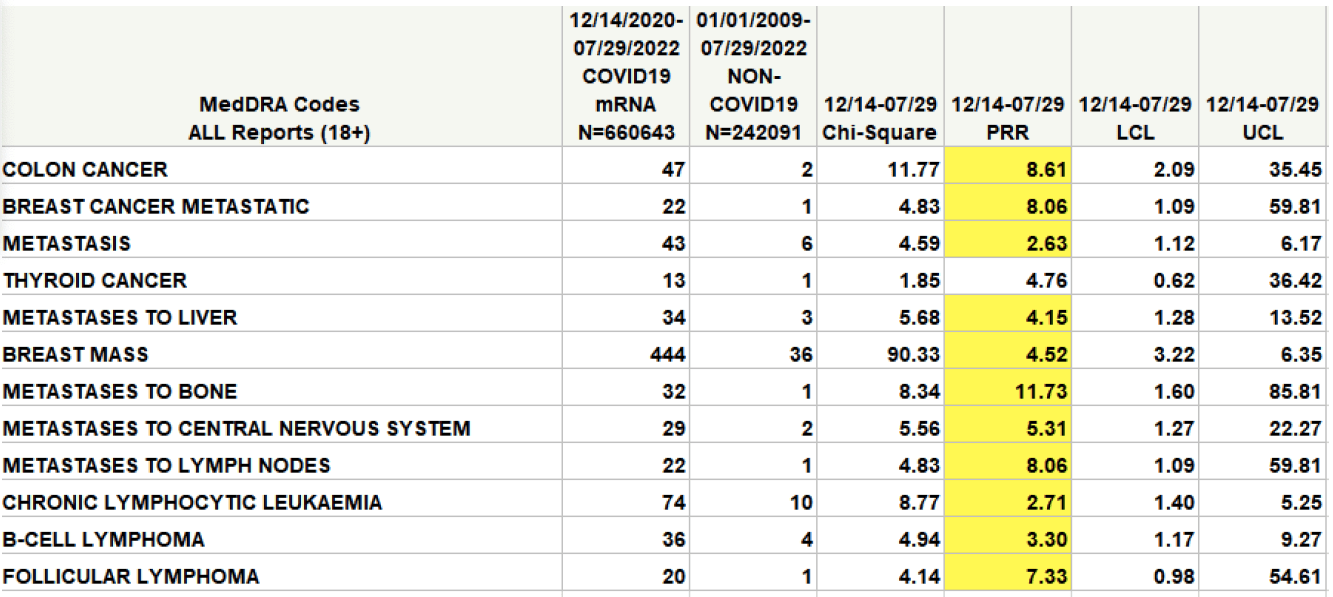
The FDA would not confirm if it found levels of DNA that exceeded acceptable levels, nor if it was investigating further.
Instead, after months of enquiries, the FDA sent boilerplate responses to me (and other media) saying, “With over a billion doses of the mRNA vaccines administered, no safety concerns related to residual DNA have been identified.”
In response to a list of questions about its testing and oversight, the FDA said it “does not have any additional information to provide at this time.”
Poor Manufacturing Oversight
We now know that Pfizer’s vaccine used in the clinical trials (PROCESS 1) was manufactured differently to the vaccine that was injected into the wider population (PROCESS 2).
This switch from PROCESS 1 to PROCESS 2 is what introduced the plasmid DNA impurities (see red circles), which could change the safety profile of the vaccine.
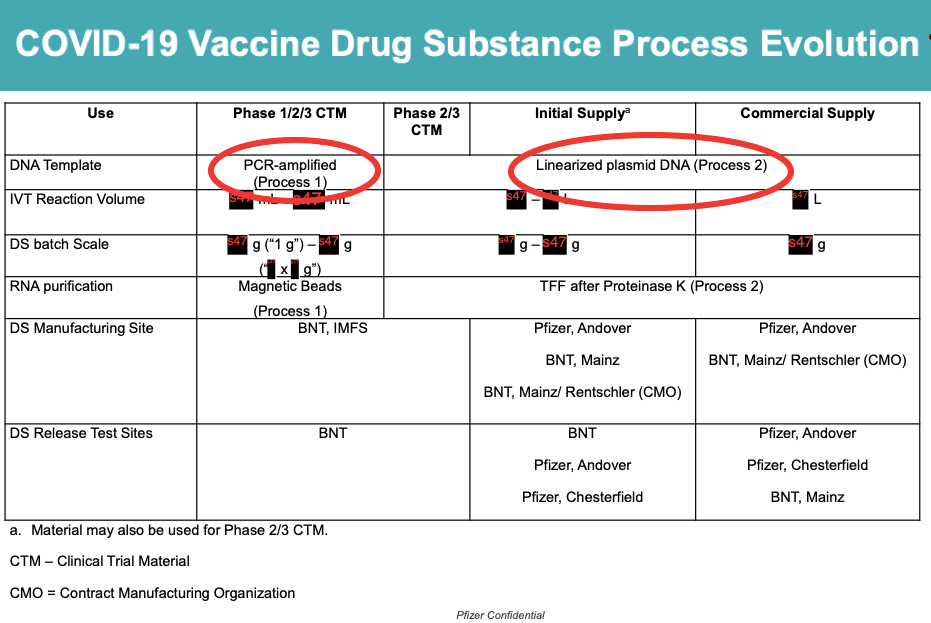
I asked the FDA if it had any human data on the comparison of the two processes.
The agency pointed me to the FDA’s EUA review memo dated 20 Nov 2020 which suggested that the testing was “ongoing.”
The three-year old document stated that “A more comprehensive comparability assessment encompassing additional lots from multiple DP manufacturing nodes is ongoing and the results will be provided to the EUA upon completion of the study.”
When I asked the FDA for access to the “ongoing” results, I was instructed to obtain the information from Pfizer, but the drug company did not respond to my enquiries.
A Freedom of Information request by Nick Hunt of the Daily Sceptic may explain why.
Pfizer promised the regulator that it would compare the safety and immunogenicity of the two processes in participants and report back by February 2021, but it seems those studies were never done.
The FOI stated:
…in October 2020 an exploratory objective was added in the C4591001 study to describe safety and immunogenicity of vaccines produced by manufacturing “Process 1” or “Process 2” in participants 16 to 55 years of age. This exploratory objective was removed and documented in protocol amendment 20 in September 2022 due to the extensive usage of vaccines manufactured via “Process 2”. Thus, this process comparison was not conducted as part of the formal documentation within the protocol amendment.[emphasis added]
Wiseman said, “Given the magnitude of the process change, from my experience in medical product development, these sorts of biological comparability studies would certainly have been expected to be undertaken by Pfizer.”
He added, “the fact that Pfizer was given a free pass indicates a significant lapse in regulatory oversight.”
Kevin McKernan, the genomics expert who made the discovery of DNA fragments in the vaccines earlier this year, says there’s “no incentive” now for Pfizer to carry out this comparative testing.
“It’s speculation on my part, but I suspect they might’ve seen increased adverse events with the commercial batch and buried the data knowing the train had left the station at that point,” said McKernan.
“There was no political will to stop vaccinating, and Pfizer probably knew the regulators would let them get away with not testing the commercial batches for the population,” he added.
Republished from the author’s Substack
Join the conversation:
Published under a Creative Commons Attribution 4.0 International License
For reprints, please set the canonical link back to the original Brownstone Institute Article and Author.








