
In part one of this article, I reviewed the contractual and regulatory framework applied by the US government to the initial development, manufacture, and acquisition of the Covid mRNA shots, using the BioNTech/Pfizer agreements to illustrate the process.
I showed that Emergency Use Authorization (EUA) was granted to these products based on clinical trials and manufacturing processes conducted with
- no binding legal standards,
- no legally proscribed safety oversight or regulation, and
- no legal redress from the manufacturer for potential harms.
In this follow-up article, I will provide a detailed analysis of the underlying documentation.
Other Transaction Authority/Agreement (OTA): A Military Acquisition Pathway
The agreement between the US government, represented by the Department of Defense (DoD), and Pfizer, representing the BioNTech/Pfizer partnership, in July 2020, for the purchase of a “vaccine to prevent COVID-19” was not an ordinary acquisition contract.
It was an agreement under Other Transaction Authority (OTA) – an acquisition pathway that, according to Department of Defense guidelines, has been used since 1958 to “permit a federal agency to enter into transactions other than contracts, grants, or cooperative agreements.”
[BOLDFACE ADDED]
A thorough review of the use of OTA by the DoD, including its statutory history, can be found in the February 22, 2019 Congressional Research Service report. This report, along with every other discussion of OTA, specifies that it is an alternative acquisition path for defense and military purposes. It is not intended, nor has it ever been used before Covid, for anything intended primarily for civilian use.
If you look for OTA laws in the US Code, this is the path you will go down:
Armed Forces -> General Military Law -> Acquisition -> Research and Engineering -> Agreements -> Authority of the DoD to carry out certain prototype projects
This legal pathway very clearly shows that OTA laws are intended for acquisition of research and engineering prototypes for the armed forces.
According to the DARPA website,
The Department of Defense has authority for three different types of OTs: (1) research OTs, (2) prototype OTs, and (3) production OTs.
These three types of OTs represent three stages of initial research, development of a prototype, and eventual production.
Within those three types, there are specific categories of projects to which OTA can apply:
- Originally, according to the OTA Overview provided by the DoD, the Other Transaction Authority was “limited to apply to weapons or weapon systems proposed to be acquired or developed by the DoD.”
- OTA was later expanded to include “any prototype project directly related to enhancing the mission effectiveness of military personnel and the supporting platforms, systems, components, or materials proposed to be acquired or developed by the DoD, or to improvement of platforms, systems, components, or materials in use by the Armed Forces.”
So far, none of that sounds like an acquisition pathway for millions of novel medical products intended primarily for civilian use.
Is There any Exception for Civilian Use of OTA That Might Apply to Covid mRNA Vaccines?
The FY2004 National Defense Authorization Act (P.L. 108-136) contained a section that gave Other Transaction Authority to “the head of an executive agency who engages in basic research, applied research, advanced research, and development projects” that “have the potential to facilitate defense against or recovery from terrorism or nuclear, biological, chemical or radiological attack.”
This provision was extended until 2018, but does not appear to have been extended beyond that year. Also, note that even in this exceptional case of non-DoD use of OTA, the situation must involve terrorism or an attack with weapons of mass destruction (CBRN).
What Other OTA Laws Might Apply?
The 2019 CRS report cited above provides this chart, showing that a few non-DoD agencies have some OTA or related authorities:
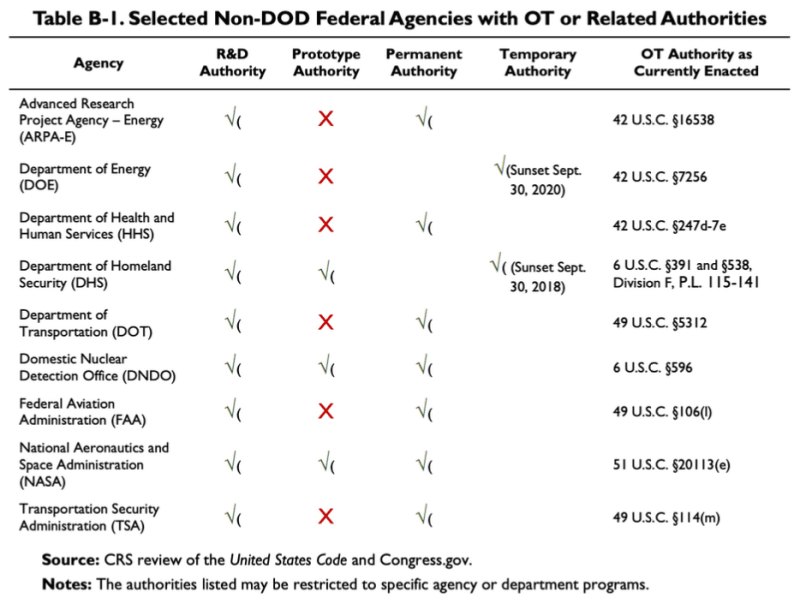
According to this table, The Department of Health and Human Services (HHS) has some research and development (R&D) Other Transaction Authorities. The law pertaining to the OT Authority of HHS is 42 U.S.C. §247d-7e.
Where is this law housed and what does it say?
The Public Health and Welfare -> Public Health Service -> General Powers and Duties -> Federal-State Cooperation -> Biomedical Advanced Research and Development Authority (BARDA) -> Transaction Authorities
So there is a place in the law related to civilian health and welfare where OTA might be applicable, although it is valid only for research and development, not prototypes or manufacturing.
The law states that the BARDA secretary has OT Authority
with respect to a product that is or may become a qualified countermeasure or a qualified pandemic or epidemic product, activities that predominantly—
(i) are conducted after basic research and preclinical development of the product; and
(ii) are related to manufacturing the product on a commercial scale and in a form that satisfies the regulatory requirements under the Federal Food, Drug, and Cosmetic Act [21 U.S.C. 301 et seq.] or under section 262 of this title.
[BOLDFACE ADDED]
The “regulatory requirements” enumerated in the law mean that it would be impossible for BARDA/HHS to enter into agreements – even just R&D – for any medical products (like the mRNA vaccines) that did not undergo rigorous safety testing and strict manufacturing oversight.
HHS “Partnership” with DoD Circumvented Civilian Protection Laws
To summarize the predicament of Other Transaction Authority/Agreements with respect to civilian authorities, in general, and Covid mRNA vaccines, in particular:
- OTA was written and codified as a way for the military to acquire weapons and other necessary systems and equipment without a lot of bureaucratic red tape. It covers research and development, prototypes, and subsequent manufacturing.
- The only OTA for a public health agency is for the HHS and it only covers Research & Development, not prototypes or manufacturing.
- Even the R&D OTA given to the HHS still requires products to be manufactured “in a form that satisfies the regulatory requirements” for drug and vaccine safety.
In other words: There is no way HHS could have used its very limited OTA to sign contracts for hundreds of millions of novel medical products.
So what did HHS do?
As the Government Accountability Office (GAO) noted in its July 2021 report on “Covid-19 Contracting:” HHS “partnered” with DoD to “leverage DoD’s OTA authorities…which HHS lacked.” (p. 24)
What are DoD’s OT Authorities for Medical Products?
As discussed, OTA is intended to help the military get equipment and technology without lots of bureaucratic hassle. None of the original laws pertaining to OTA mentioned anything other than “platforms, systems, components, or materials” intended to “enhance the mission effectiveness of military personnel.”
But five years before Covid, an exceptional use of OTA was introduced:
In 2015, DoD announced the establishment of the CBRN Medical Countermeasure Consortium, whose purpose was to use the OTA acquisition pathway to “work with DoD to develop FDA licensed chemical, biological, radiological, and nuclear medical countermeasures.” [FDA = Food & Drug Administration]
As described in the 2015 announcement, this included “prototype technologies for therapeutic medical countermeasures targeting viral, bacterial, and biological toxin targets of interest to the DoD.” The list of agents included the top biowarfare pathogens, such as anthrax, ebola, and marburg.
The announcement went on to specify that “enabling technologies can include animal models of viral, bacterial or biological toxin disease and pathogenesis (multiple routes of exposure), assays, diagnostic technologies or other platform technologies that can be applied to development of approved or licensed MCMs [medical countermeasures].”
Although this still does not sound anything like the production of 100 million novel vaccines for civilian use, it does provide more leeway for OTA than the very limited Other Transaction Authority given to HHS.
While the HHS OTA requires adherence to extensive development and manufacturing regulations, the OTA pathway for the DoD to develop medical countermeasures requires only “FDA licensure.”
Thus, using DoD Other Transaction Authorities, it would theoretically be possible to bypass any safety regulations – depending on the requirements for FDA licensing of an OTA-generated product. As we will see, in the case of the Covid mRNA vaccines, Emergency Use Authorization was granted, requiring no legal safety oversight at all.
Emergency Use Authorization (EUA)
Here’s how the Food & Drug Administration (FDA) describes its EUA powers:
Section 564 of the FD&C Act (21 U.S.C. 360bbb–3) allows FDA to strengthen public health protections against biological, chemical, nuclear, and radiological agents.
With this EUA authority, FDA can help ensure that medical countermeasures may be used in emergencies to diagnose, treat, or prevent serious or life-threatening diseases or conditions caused by biological, chemical, nuclear, or radiological agents when there are no adequate, approved, and available alternatives (among other criteria).
It’s extremely important to understand that these EUA powers were granted in 2004 under very specific circumstances related to preparedness for attacks by weapons of mass destruction, otherwise known as CBRN (chemical, biological, radiological, nuclear) agents.
As explained in Harvard Law’s Bill of Health,
Ultimately, it was the War on Terror that would give rise to emergency use authorization. After the events of September 11, 2001 and subsequent anthrax mail attacks, Congress enacted the Project Bioshield Act of 2004. The act called for billions of dollars in appropriations for purchasing vaccines in preparation for a bioterror attack, and for stockpiling of emergency countermeasures. To be able to act rapidly in an emergency, Congress allowed FDA to authorize formally unapproved products for emergency use against a threat to public health and safety (subject to a declaration of emergency by HHS). The record indicates that Congress was focused on the threat of bioterror specifically, not on preparing for a naturally-occurring pandemic.
The wording of the EUA law underscores the fact that it was intended for use in situations involving weapons of mass destruction. Here are the 4 situations in which EUA can be issued:
- a determination by the Secretary of Homeland Security that there is a domestic emergency, or a significant potential for a domestic emergency, involving a heightened risk of attack with a biological, chemical, radiological, or nuclear agent or agents;
- a determination by the Secretary of Defense that there is a military emergency, or a significant potential for a military emergency, involving a heightened risk to United States military forces, including personnel operating under the authority of Title 10 or Title 50, of attack with—
- a biological, chemical, radiological, or nuclear agent or agents; or
- an agent or agents that may cause, or are otherwise associated with, an imminently life-threatening and specific risk to United States military forces;
- a determination by the Secretary that there is a public health emergency, or a significant potential for a public health emergency, that affects, or has a significant potential to affect, national security or the health and security of United States citizens living abroad, and that involves a biological, chemical, radiological, or nuclear agent or agents, or a disease or condition that may be attributable to such agent or agents; or
- the identification of a material threat pursuant to section 319F–2 of the Public Health Service Act [42 U.S.C. 247d–6b] sufficient to affect national security or the health and security of United States citizens living abroad.
Nowhere in these four situations is there any mention of a naturally occurring epidemic, pandemic, or any other kind of public health situation that is not caused by “biological, chemical, radiological or nuclear agent/s.”
Could SARS-CoV-2 qualify as such an agent?
If you look for the definition of “biological agents” in the US Legal Code, you will go down the following pathway:
Crimes and Criminal Procedure -> Crimes -> Biological Weapons -> Definitions
So in the context of United States law, the term “biological agents” means biological weapons, and the use of such agents/weapons is regarded as a crime.
Wikipedia provides this definition:
A biological agent (also called bio-agent, biological threat agent, biological warfare agent, biological weapon, or bioweapon) is a bacterium, virus, protozoan, parasite, fungus, or toxin that can be used purposefully as a weapon in bioterrorism or biological warfare (BW).
On What Legal Basis was EUA Issued for Covid mRNA Vaccines?
It would seem, based on the laws regarding EUA, that none of the four possible situations described in the law could be applied to a product intended to prevent or treat a disease caused by a naturally occurring pathogen.
Nevertheless, this law was used to authorize the mRNA Covid vaccines.
Given the four choices listed in the EUA law, the one that was used for Covid “countermeasures” was
C) a determination by the Secretary that there is a public health emergency, or a significant potential for a public health emergency, that affects, or has a significant potential to affect, national security or the health and security of United States citizens living abroad, and that involves a biological, chemical, radiological, or nuclear agent or agents, or a disease or condition that may be attributable to such agent or agents.
When applied specifically to Covid, this is how it was worded:
the Secretary of the Department of Health and Human Services (HHS) determined that there is a public health emergency that has a significant potential to affect national security or the health and security of United States citizens living abroad, and that involves the virus that causes Coronavirus Disease 2019 (COVID-19)…
There is no doubt here that “the virus that causes COVID-19” is deemed to be the equivalent of “a biological, chemical, radiological, or nuclear agent or agents.”
It is also important to note that the EUA “determination of a public health emergency” is completely separate from, and not in any way reliant on, any other public health emergency declarations, like the ones that were made by the WHO, the US government, and the President at the beginning of the Covid-19 pandemic.
So even when the WHO, the US government, and the President declare that the pandemic is over, there can still be Emergency Use Authorization if the HHS Secretary continues to claim that the situation described in section C) exists.
Looking at all of the EUAs for hundreds of Covid-related medical products, it is very difficult to see how the HHS secretary could justify the claim that “there is a public health emergency that has a significant potential to affect national security or the health and security of US citizens living abroad” in most, if not all, of these cases.
Additional “Statutory Criteria” for FDA to Grant Emergency Use Authorization
Once the HHS Secretary declares that there is a public health emergency that warrants EUA, based on one of the four situations listed in the law, there are four more “statutory criteria” that have to be met in order for the FDA to issue the EUA. Here’s how the FDA explains these requirements:
- Serious or Life-Threatening Disease or Condition
For FDA to issue an EUA, the CBRN agent(s) referred to in the HHS Secretary’s EUA declaration must be capable of causing a serious or life-threatening disease or condition.
NOTE: This criterion repeats the specification of a CBRN agent, which is legally defined as a weapon used in committing a crime.
- Evidence of Effectiveness
Medical products that may be considered for an EUA are those that “may be effective” to prevent, diagnose, or treat serious or life-threatening diseases or conditions that can be caused by a CBRN agent(s) identified in the HHS Secretary’s declaration of emergency or threat of emergency under section 564(b).
The “may be effective” standard for EUAs provides for a lower level of evidence than the “effectiveness” standard that FDA uses for product approvals. FDA intends to assess the potential effectiveness of a possible EUA product on a case-by-case basis using a risk-benefit analysis, as explained below.
[BOLDFACE ADDED]
LEGAL QUESTION: How can anyone legally claim that a product authorized under EUA is “safe and effective” if the legal standard for EUA is “may be effective” and the FDA declares that this is a “lower level of evidence” than the standard used for regular product approvals?
- Risk-Benefit Analysis
A product may be considered for an EUA if the Commissioner determines that the known and potential benefits of the product, when used to diagnose, prevent, or treat the identified disease or condition, outweigh the known and potential risks of the product.
In determining whether the known and potential benefits of the product outweigh the known and potential risks, FDA intends to look at the totality of the scientific evidence to make an overall risk-benefit determination. Such evidence, which could arise from a variety of sources, may include (but is not limited to): results of domestic and foreign clinical trials, in vivo efficacy data from animal models, and in vitro data, available for FDA consideration. FDA will also assess the quality and quantity of the available evidence, given the current state of scientific knowledge.
[BOLDFACE ADDED]
LEGAL NOTE: There is no legal standard and there are no legal definitions for what it means for “known and potential benefits” to outweigh “known and potential risks.” There is also no qualitative or quantitative legal definition for what constitutes acceptable “available evidence” upon which the risk-benefit analysis “may be” based. There could be zero actual evidence, but a belief that a product has a lot of potential benefit and not a lot of potential risk, and that would satisfy this “statutory requirement.”
- No Alternatives
For FDA to issue an EUA, there must be no adequate, approved, and available alternative to the candidate product for diagnosing, preventing, or treating the disease or condition. A potential alternative product may be considered “unavailable” if there are insufficient supplies of the approved alternative to fully meet the emergency need.
LEGAL QUERY: Aside from the egregious and potentially criminal vilification/outlawing of alternative Covid-19 treatments like ivermectin and hydroxychloroquine, at what point was there an approved alternative for “preventing Covid-19” (the only thing the mRNA vaccines were purchased to do) – Paxlovid, for instance – which would render an EUA for the mRNA vaccines no longer legal?
Here’s how all of these “statutory criteria” were satisfied in the actual Emergency Use Authorization for the BioNTEch/Pfizer Covid mRNA vaccines:
I have concluded that the emergency use of Pfizer-BioNTech COVID‑19 Vaccine for the prevention of COVID-19 when administered as described in the Scope of Authorization (Section II) meets the criteria for issuance of an authorization under Section 564(c) of the Act, because:
- SARS-CoV-2 can cause a serious or life-threatening disease or condition, including severe respiratory illness, to humans infected by this virus;
- Based on the totality of scientific evidence available to FDA, it is reasonable to believe that Pfizer-BioNTech COVID‑19 Vaccine may be effective in preventing COVID-19, and that, when used under the conditions described in this authorization, the known and potential benefits of Pfizer-BioNTech COVID‑19 Vaccine when used to prevent COVID-19 outweigh its known and potential risks; and
- There is no adequate, approved, and available alternative to the emergency use of Pfizer-BioNTech COVID‑19 Vaccine to prevent COVID-19.
[BOLDFACE ADDED]
NOTE: The only context in which the FDA weighed the potential benefits and risks of the vaccine, and in which the FDA determined it “may be effective” was in preventing Covid-19.
There is no consideration, no evidence of actual or potential benefit, and no determination that there is any potential effectiveness for the vaccine to do anything else, including: lowering the risk of severe disease, lowering the risk of hospitalization, lowering the risk of death, lowering the risk of any conditions actually or potentially related to Covid-19.
THEREFORE, one might reasonably question the legality of any claims that the vaccine is “safe and effective” in the context of anything other than “when used to prevent COVID-19” – which the vaccines were known NOT TO DO very soon after they were introduced.
If people were told the BioNTech/Pfizer mRNA vaccines were “safe and effective” at anything other than preventing Covid-19, and if they were threatened with any consequences for failure to take the vaccine for anything other than preventing Covid-19, might they have a legitimate argument that they were illegally coerced into taking an unapproved product under fraudulent claims?
Third-Tier Requirements for EUA for Unapproved Products
Once we have the EUA-specific emergency declaration, and once the FDA declares that the product may be effective and that whatever evidence is available (from zero to infinity) shows that its benefits outweigh its risks (as determined by whatever the FDA thinks those might be), there is one more layer of non-safety, non-efficacy related regulation.
Here’s how a 2018 Congressional Research Service report on EUA explains this:
FFDCA §564 directs FDA to impose certain required conditions in an EUA and allows for additional discretionary conditions where appropriate. The required conditions vary depending upon whether the EUA is for an unapproved product or for an unapproved use of an approved product. For an unapproved product, the conditions of use must:
(1) ensure that health care professionals administering the product receive required information;
(2) ensure that individuals to whom the product is administered receive required information;
(3) provide for the monitoring and reporting of adverse events associated with the product; and
(4) provide for record-keeping and reporting by the manufacturer.
LEGAL QUESTION: What exactly is the “required information?” We know that people were informed that the vaccines were given Emergency Use Authorization. But were they told that this means “a lower level of evidence” than is required for “safe and effective” claims on other medical products? Were they informed that there are different levels of “safe and effective” depending on whether a product has EUA or another type of authorization?
NOTE: The law requires that there be a way to monitor and report adverse events. However, it does not state who monitors, what the standards are for reporting, and what the threshold is for taking action based on the reports.
EUA Compared to Every Other Drug/Vaccines Approval Pathway
As researcher/writer Sasha Latypova has pointed out, many people were confused by EUA, because it sounds a lot like EAU, which stands for “Expanded Access Use.” This is a type of authorization given to medical products when there is urgent need by a particular group of patients (e.g., Stage IV cancer patients whose life expectancy is measured in months) who are willing to risk adverse events and even death in exchange for access to an experimental treatment.
Emergency Use Authorization is in no way related to, nor does it bear any resemblance to, Expanded Access Use.
The various legal pathways for authorizing medical products are neatly presented in a table highlighted by legal researcher Katherine Watt. The table is part of a 2020 presentation for an FDA-CDC Joint Learning Session: Regulatory Updates on Use of Medical Countermeasures.
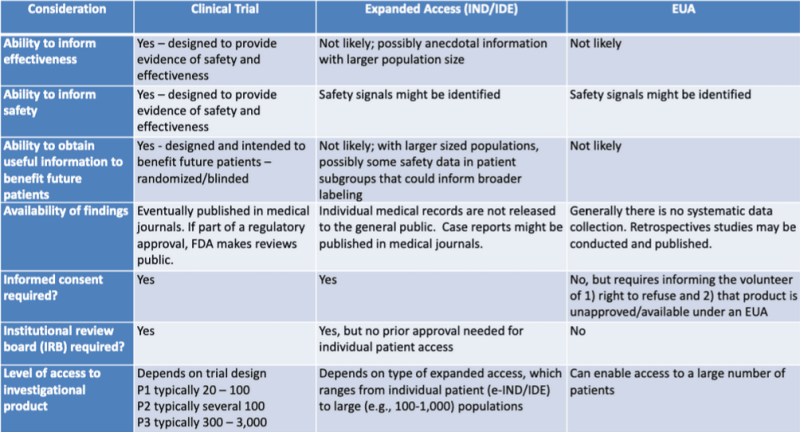
This table shows very clearly that the EUA process is unlikely to provide information regarding product effectiveness, is not designed to provide evidence of safety, is not likely to provide useful information to benefit future patients, involves no systematic data collection, requires no retrospective studies, no informed consent, and no institutional review board.
Moreover, in a 2009 Institute of Medicine of the National Academic publication, also highlighted by Watt, entitled “Medical Countermeasures: Dispensing Emergency Use Authorization and the Postal Model – Workshop Summary” we find this statement on p. 28:
It is important to recognize that an EUA is not part of the development pathway; it is an entirely separate entity that is used only during emergency situations and is not part of the drug approval process.
Does this mean that approvals of Covid-19 countermeasures that were based on EUAs were illegal? Does it mean that there is no legal way to claim an EUA product is “safe and effective” because it is NOT PART OF THE DRUG APPROVAL PROCESS?
Conclusion
It is eminently apparent, given all the information in this article, and in the preceding Part 1, that the BioNTach/Pfizer Covid mRNA vaccines were developed, manufactured, and authorized under military laws reserved for emergency situations involving biological warfare/terrorism, not naturally occurring diseases affecting the entire civilian population.
Therefore, the adherence to regulations and oversight that we expect to find when a product is deemed “safe and effective” for the entire civilian population was not legally required.
Can this analysis be used to challenge the legality of the “safe and effective” claim by those government officials who knew what EUA entailed? Are there other legal ramifications?
I hope so.
Importantly, in legal challenges to Covid mRNA vaccines brought so far, there have been no rulings (that I am aware of) on whether military law, like OTA and EUA, can be applied to civilian situations. However, there has been a statement by District Court Judge Michael Truncale, in his dismissal of the case of whistleblower Brook Jackson v. Ventavia and Pfizer, that is important to keep in mind.
Here the judge acknowledges that the agreement for the BioNTech/Pfizer mRNA vaccines was a military OTA, but he refuses to rule on its applicability to the non-military circumstances (naturally occurring disease, 100 million doses mostly not for military use) under which it was issued:
The fact that both military personnel and civilians received the vaccine does not indicate that acquiring the vaccine was irrelevant to enhancing the military’s mission effectiveness. More importantly, Ms. Jackson is in effect asking this Court to overrule the DoD’s decision to exercise Other Transaction Authority to purchase Pfizer’s vaccine. But as the United States Supreme Court has long emphasized, the “complex subtle, and professional decisions as to the composition, training, equipping, and control of a military force are essentially professional military judgments.” Gilligan v. Morgan, 413 U.S. 1, 10 (1973). Thus, it is “difficult to conceive of an area of governmental activity in which the courts have less competence.” Id. This Court will not veto the DoD’s judgments concerning mission effectiveness during a national emergency.
This is just one of many legal hurdles that remain in the battle to ultimately outlaw all mRNA products approved during the Covid-19 emergency, and any subsequent mRNA products whose approval was based on the Covid-19 approval process.
Join the Conversation
Published under a Creative Commons Attribution 4.0 International License
For reprints, please set the canonical link back to the original Brownstone Institute Article and Author.








