Quality control errors during pharmaceutical manufacturing are unfortunately common occurrences. The majority (around 50%) of employees that work in pharmaceutical manufacturing tend to have duties which are safety/quality-control related, but apparently even that isn’t enough.
Even with a major focus on quality, there is still an estimated 2-3σ (sigma) level of imprecision when it comes to pharmaceutical manufacturing. That corresponds to 66,807 to 308,537 defects per 1,000,000 opportunities. But with pharmaceutical development being so complicated, there could be more than 1,000,000 “opportunities” for error.
The above listed error calculation – while alarming enough – was referenced in small-molecule pharmacology. However, increasingly complex pharmaceuticals (such as today’s widely used biotechnological products, including GLP-1 diabetes/weight loss or mRNA products for Covid-19) have molecular weights that can be thousands times larger than small molecule compounds. That could mean an even greater opportunity for error.
The FDA is abundantly aware of pharmaceutical fragility and potential quality shortcomings, including at the highest levels of its leadership.
In fact, Dr. Michael Kopcha, the current Director of the FDA’s Office of Pharmaceutical Quality (OPQ), wrote and published the above published Six Sigma calculation, lamenting the imprecise nature of pharmaceutical manufacturing – back in 2017.
Any alteration in structure that occurs during manufacturing has the potential to vastly change a compound’s clinical activity, including a change from therapeutic drug into a poison.
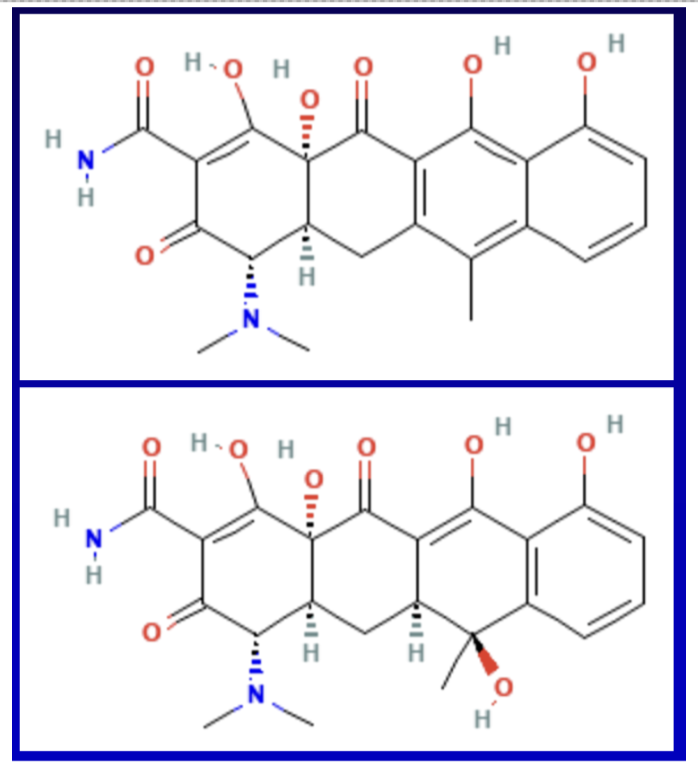
For instance, tetracycline illustrated above has a molecular weight of approximately 445g/mol. Covid mRNA vaccines are estimated to be 3,000 times greater in size than tetracycline, not including the substantial, varied, and clinically complex lipid nanoparticles.
Larger structures mean more potential for error. It is plausible that mRNA product quality control deviations could be the reason that hundreds of thousands of Americans died or have gotten severely ill from Covid injections, while other Americans have had minor or no adverse events reported. It’s an especially critical matter because in addition to being highly complex, Covid treatments were jammed through development, authorization, approval, and manufacturing at “warp speed.” Finally, neither manufacturers nor the FDA are disclosing the exact structure or ingredients of Covid mRNA “vaccines,” which means that the public is in the blind and 100% dependent on the FDA to independently collect and test Covid mRNA shots for quality control.
While this piece focuses on drugs that the OPQ regulates, a “Part 2” to this article will address things like pet foods, human foods, baby formula, nicotine/e-liquids, supplements, and everything else that isn’t the “D” part of the FDA.
Rampant Fraud ON TOP OF Poor Quality from Chinese and Indian Drug Manufacturers:
Low prices on imported products from China and India come with a different kind of cost…that of quality. And there is a big difference between buying an inexpensive or throwaway Chinese-made LED/USB gadget for a couple of dollars, versus swallowing pills of nebulous quality, every day for months, years, or decades.
China and India are where America gets the vast majority of its generic drug supply. Both countries have both historic and recent histories of poor quality control, on top of egregious instances of pharmaceutical fraud.
These are not recent, rare, or outlier occurrences; fraud from China and India has been documented over decades and in great detail within FDA inspection report archives. For example, Ranbaxy, a (now defunct) generic drug manufacturing plant in India, was fined $500 million for repeatedly lying to the FDA about testing drugs for quality and consistency. Some of the Ranbaxy drugs included those used to treat vulnerable HIV/AIDS patients, where both timing and precision dosing of agents are particularly critical and likely directly affected patient outcomes. Generally speaking, there exists a decade-plus history of fraud and data falsification when it comes to pharmaceutical supply from China and India, coinciding with when pharmaceutical manufacturers began relocating there.
FDA inspection reports detail a remarkable disdain for the most basic regulatory standards for manufacturing, quality, recordkeeping, and even basic standards for cleanliness – for products that overseas manufacturers like China and India fully realize are destined to the United States and other Western nations.
Despite legendary poor quality, profit-centric pharmacies across the USA, including CVS and Walgreens, appear mostly beguiled with acquiring its inventory at the lowest possible prices; ie, from Chinese and Indian manufacturers.
With a track record and pervasiveness of errors and fraud, one would think that the FDA would be exceptionally vigilant about testing everything coming in from those countries, but they would be wrong.
Or to more aptly put it – dead wrong.
Incomplete FDA Oversight Means Poor Quality Drugs at American Hospitals and Pharmacies:
Since manufacturing is highly complicated and attention to quality so critical, a legitimate question to raise would be: Who is assuring the quality of our pharmaceuticals and how are they doing so?
Most people think that the FDA independently collects and verifies the purity of what China and India sell to America’s pharmacies. However, according to official FDA records and publications, the FDA doesn’t independently collect and test >99.9% of drugs from overseas, including China and India.
Additionally, the specific products that the FDA chooses to test, and the methodology employed, raises questions.
2021, 2022 & 2023 FDA OPQ Testing:
- According to their website, for all of 2021, the FDA’s OPQ only independently collected and conducted quality control on a mere 137 (non-unique) drugs. 73 of 137 (53%) of those products selected were “hand sanitizers.” Only five different active ingredients that the OPQ selected for testing were top 100 prescribed drugs in the US.
- For all of 2022, the FDA’s OPQ only tested 376 non-unique products. 278 of 376 (74%) of those products were “hand sanitizers.” None of the active ingredients tested were top-100 prescribed drugs in the US.
- For all of 2023, the FDA’s OPQ only tested 79 (non-unique) products. Only three different active ingredients were top-100 prescribed drugs in the US.
- No information about anything that the FDA tested in 2024 is currently available, despite this article being written in late October 2024.
Does this mean that of the tens of thousands of pharmaceutical products that the FDA regulates, for 2021, 2022 and 2023, almost exclusively chose to conduct quality control testing…on hand sanitizers??
According to publicly available data from the FDA’s OPQ, that answer is YES.
The disproportionate testing of “hand sanitizers” and/or obscure items as some of the very few products that the FDA independently collected and tested isn’t something new for 2022. As can be seen in the linked spreadsheets, the FDA did the same back in 2020 and 2021 as well.
At the same time, the FDA largely omitted high-volume, top-100 drugs not to mention federally mandated, highly complex Covid mRNA injections despite them being manufactured at “warp speed.” Did the FDA independently collect and test mandated mRNA “vaccines?” Today those injections are associated with over ONE MILLION adverse events which have the potential to be directly related to the drug quality/product consistency deviations.
Questions about the Very Few Products That the FDA’s OPQ Does Test:
Critical issues and questions about drug testing include:
- Who or what FDA personnel or scientific methodology determined that independently collecting and testing a mere ~0.001% of drugs is an appropriate representation of all 150,000 products that OPQ oversees?
- Delaying releasing quality control data by the OPQ for around a year (ie, 2023 data was not released until around March of 2024, and none of the 2024 OPQ findings were shared, even in late October) does almost nothing to help America’s patients. That sort of delay does nothing to help Americans wanting to look up testing information about the quality of their drugs they are taking right now.
- OPQ’s data minimally disclosed information about “Tests Performed” only listed things like “Friability/tablet hardness” and “dissolution” and “aluminum” and “visual examination…” Did the OPQ not perform thorough pharmaceutical release testing to check for things like milligram accuracy, or bacterial/microbial contamination? Why not conduct full testing and share complete results?
- The “Results” columns only provided highly simplified and vague explanations such as “Passed (tests)” or “Failed (tests)” or “Failed impurities” providing no specific information on percentage purity, lot numbers, units per lot reaching American consumers, methodology employed, instruments used, calibration, or tolerances. It additionally lacked exactly naming what the specific impurities were, or the degree of the impurity (eg, was impurity 1 part per 1,000 vs 1:100,000 vs 1:1,000,000 vs 1:1,000,000,000?).
- Typical analyses provide comprehensive quantitative and qualitative and other analytical findings and/or should include a full explanation of potential implications for what the failed test could mean clinically – especially because it is the FDA that is conducting it – and especially when those products are being given to already sick, and/or hospitalized – whose life is on the line.
- Instead of burying OPQ results in a drop-down menu at the bottom of a webpage that is not linked anywhere on the FDA homepage or anywhere that most consumers and clinicians could ever find it, why not list results more prominently and/or share a press release on both positive and negative findings?
- Another significant OPQ failure included a lack of detailed follow-up instructions for patients to follow if they somehow managed to discover that they had taken a product of poor quality.
Those include:- What should patients do? Do patients need to speak with their pharmacist, or physician? What information do they need to tell them? Will insurances cover these claims or are patients expected to pay out-of-pocket for replacement medication and physician visits?
- Should patients immediately stop taking their medication which does not pass FDA OPQ analytical testing?
- Pharmacists and physicians are not toxicologists. Why won’t the FDA’s OPQ or medical officers provide specific medical or laboratory tests alongside failed test results and/or other specific guidance?
- Why is there no telephone or email for patients/pharmacists/physicians to contact the FDA’s OPQ for specific recommendations? Are patients/pharmacists/physicians supposed to figure it out on their own?
- Should patients contact a poison control center? Will they have adequate knowledge on what to do?
- What attempts were made by the FDA/manufacturers to contact affected patients to inform them of failed tests, since failed quality testing results are not posted on their website in a timely manner?
- Was there any manufacturer or specific employee punishment or other accountability for out-of-spec products? If not, why not?
- What was the cascade of events that led to the errant findings and what policies/procedures were amended by the FDA and/or manufacturers to prevent the same error(s) from occurring again?
Since it is difficult to navigate through the FDA website (perhaps intentionally?) to find the products that were tested, and since FDA webpages have a mysterious habit of “disappearing” after a while, (anyone checking out my earlier writings will find my articles filled with now dead links to FDA.gov websites), I am linking a direct download to an Excel spreadsheet of what was tested and what results were provided by the FDA.
The FDA Targeting Small American Businesses with Quality Control Testing and Inspections?
Of the supremely few drugs that the FDA does choose to test, the FDA disproportionately chose products that are made in the US, despite the fact that almost all pharmaceutical manufacturing takes place overseas.
For instance, in 2021, Rock Town Distillery, a small business in Little Rock, Arkansas, was one of hundreds of US-based manufacturers that had its ethanol-based hand sanitizer analyzed by the FDA’s OPQ for quality.
The incongruent focus of the FDA on the few domestic small business manufacturers versus multibillion dollar manufacturers in China and India and the odd choice of “hand sanitizers” out of the 150,000 total products the FDA’s OPQ regulates is questionable to say the least.
It is especially upsetting because Rock Town Distillery volunteered to give away their hand sanitizer for free during Covid. And they did so as a response to Donald Trump’s Presidential Defense Production Act to produce “essential medical countermeasures” for Covid to help their fellow countrymen.
Rock Town Distillery – which is a small business bar/distillery – grosses less than $5 million per year and has less than 10 employees. Compare that to Cipla, an Indian pharmaceutical company, which has a revenue of $3 billion per year, a market cap of over $13.5 billion and over 26,000 employees, and sells about 1,500 products in 86 different countries.
While the FDA inspected and analyzed hundreds of small businesses like Rock Town Distillery for their “hand sanitizers,” I could find NO evidence of Pfizer or Moderna or any other Big Pharma Covid drug manufacturers being inspected or analyzed for quality control by the OPQ – or by other division at the FDA – on their highly-complex “warp speed” manufactured products.
Rock Town Distillery was far from alone. In fact: approximately 300 small, domestic “mom-and-pop” American businesses and micro-distilleries had audits by the FDA’s OPQ for quality control testing. At the same time, the FDA seemed to give a pass to massive, overseas, multibillion-dollar manufacturing facilities in China and India.
Is the FDA’s disproportionate focus on US-based distilleries an example of political persecution by the “nonpartisan” FDA against small businesses who supported Trump’s Pandemic Countermeasure Order? See the linked yearly spreadsheet/tabs for everything that was tested over the past four years and judge for yourself!
The disproportionate focus of the FDA on American small businesses seems biased. It appears to illustrate yet another instance in what has become a long list of examples of an explosion of the growth of Wall Street built on the destruction of Main Street (ie, small businesses).
Is the FDA now “in” on further bolstering big, wealthy corporations by targeting small business for ruin? It’s generally happening elsewhere. Under the Biden/Harris administration, there are 37.5% fewer small businesses open nationwide compared with January 2020, two months before the pandemic hit the United States.


Why are the FDA’s OPQ Directors Dr. Michael Kopcha and/or Dr. Jennifer Maguire disproportionately focusing their quality auditing of small American businesses that volunteered to manufacture essential medical countermeasures to the Covid-19 pandemic as some sort of awful partisan political revenge? In light of what they chose to test these past few years, it’s not an absurd question to raise.
Dr. Maguire posts on Twitter about the work she does at the FDA under the name “@Quality_Sleuth.”
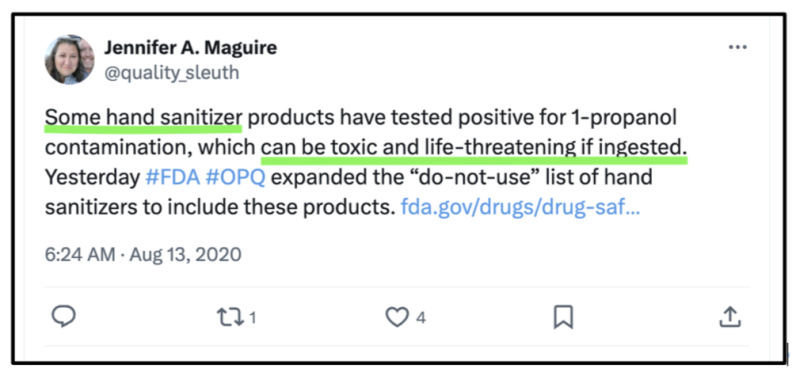
Apparently, “hand sanitizers” were singled out because “some” hand sanitizer products had tested positive for 1-propanol, which according to Dr. Maguire “…can be toxic and life-threatening if ingested.”
Of course “hand sanitizers” are only intended to be used on one’s… “hands”…not ingested!
Arguably, even if perfectly made hand sanitizers were ingested, they could be expected to produce untoward and/or toxic effects. Despite that, they received heavily disproportionate OPQ scrutiny during both 2021 and 2022.
Arguably, any regulated product placed under the same levels of far-fetched scrutiny, and/or if obviously misused (ie, oral ingestion of a topical product) would be the cause of similar safety/adverse event concerns.
For example: how many ingredients that make up orally-administered, say, blood pressure pills that if crushed or broken are eye irritants? Answer: Probably most of them. Does that mean the FDA’s OPQ office has cause to start auditing all Chinese-and Indian-made blood pressure medications for impurities? How and why was hand sanitizer ingestion chosen as a focus?
According to PubChem, the topical safety and toxicity risks of 1-propanol are similar to that of ethanol, isopropyl alcohol, or benzalkonium chloride, each of which are individually used as the main active antiseptic ingredients of hand sanitizers. Furthermore, a search to the FDA Adverse Event Database (AERS) website for 1-propanol reports showed only four reports in the 2020-to-latest available AERS database, which according to the FDA are “unverified” and are “not definitive proof of the causal relationship between exposure to the product and the reported event.”
While hand sanitizers should accurately list labeled ingredients (and not have contained 1-propanol unless it met marketing requirements under FDCA 505-G), was the pathologic ingestion of “hand sanitizers” by a fringe population of four individuals across the world enough cause for it to be 53% and 74% focus of what the FDA’s OPQ 1,300 employees tested for 2021 and 2022 respectively?
Why was it prioritized over collecting and testing high-use, top-100 drugs made by Chinese and Indian pharmaceutical companies?
These are questions for which the American public deserves an explanation and complete transparency about.
The FDA Isn’t Keeping up with the Drug Manufacturing Pilgrimage to China and India:
Hand sanitizers notwithstanding, in case one thought 2021 – 2023 testing data were outliers –with regards to the very few products tested and/or a focus on US-made products – they’re not. The FDA’s OPQ sampling website shows a heavily disproportionate preference to test domestically made products versus those made overseas, according to data based on what the FDA’s OPQ has selected to test from 2013 to 2023.
That’s despite the fact that during the same time interval, most brand-name drugs (and around 85% of generic drugs) were manufactured overseas.
According to the website www.fda483s.com, despite the fact that the overwhelming volume of pharmaceutical manufacturing occurs in China and India, during 2013 to 2019, only 243 of the FDA’s 2,344 violations (ie, approximately 10%) were issued to facilities in China and India.
(Note: the FDA Form 483 is a form issued to a facility to notify them of various deficiencies/violations following a live, in-person inspection, which is what the fda483s.com website catalogs and tracks.)
It’s certainly not because those Chinese and Indian facilities are regarded for producing superior quality products – it’s because the FDA only rarely conducts live, in-person inspections or end-user testing on overseas-manufactured products. For well over a decade, the FDA instead appeared to focus its inspections on domestic manufacturers of FDA-regulated products.
One reason for that could be: international travel and the accompanying State-Department clearance and international communications to foreign governments, various permissions paperwork, visas, security, language barriers, and other coordination needed for federal employees to travel overseas to China and India on official business. International requirements are substantially more arduous and time-intensive than simply traveling within the United States by car or airplane to perform inspection at a domestic facility, requiring comparatively little paperwork and no international coordination with governments and the State Department.
For these reasons, both independent collection and testing as well as inspections seem to have mainly focused on the United States. And although Covid made it worse, that has been going on for over a decade.
In fact, motivation for manufacturers to relocate to China/India – may not just only be to 1) lower labor costs, but to also 2) escape the FDA’s oversight via impromptu and more frequent domestic inspections. While overseas facilities are notified in advance of inspections via the required State Department protocol notification processes listed above, domestic facilities receive NO notification and no opportunities to prepare.
Despite the pharmaceutical manufacturing pilgrimage overseas, a Bloomberg article of a ProPublica analysis of FDA inspection data showed that the number of the agency’s inspections of drug manufacturers in India and China actually dropped.
Quoting an excerpt from the article:
In fiscal year 2019, the year before the COVID-19 pandemic limited travel and movement, the FDA inspected 37% of the nearly 2,500 overseas manufacturers; in 2022, the [FDA] only inspected 6% of around 2,800. And in India… the FDA inspected only 3% of manufacturers in 2022. (emphases added)
Then, following the pandemic, even those few remaining in-person overseas inspections completely halted – despite the fact that the FDA itself had authorized and/or approved “safe and effective” mRNA injections for Covid.
In 2021, the FDA then proposed and advanced a new and preposterous policy to replace inspections completely.
Starting Under Biden/Harris, Quality Control Samples from China and India Are “Mailed In”
Back in January 2021, and almost immediately after President Biden assumed office, the FDA decided to start monitoring America’s drug quality via a remote collection of manufacturer-supplied “mailed-in” sampling seemingly as a replacement for conventional, in-person, collections during facility inspections.
One of the justifications made by the FDA for “mailed-in” sampling in lieu of live, in-person collections was Covid, despite there (ironically) being an FDA-advertised “safe and effective mRNA vaccine” available in late 2020.


mRNA shot efficacy notwithstanding, even after it was shown that morbidity and mortality collapsed following emergence of the Omicron variant in 2021, the FDA continued to advance a permanent “mailed-in” sampling methodology agency-wide.
Asking manufacturers to submit “mail-in” samples for quality testing obviously leaves vulnerability for fraud – especially when it entrusts that methodology to countries like China and India – both of which have long histories of fraud.
It’s as unacceptable and absurd as if, for instance, state health inspectors – as an alternative to scrutinizing restaurants in-person – asked restaurant owners to “mail in” entrées for testing.
As is obvious, restaurant owners (or drug manufacturers) can curate and customize what samples they prefer to submit as representative of larger batches.
That concern is compounded when one notes that a large fraction of pharmaceutical manufacturers in China are state-owned, with additional private Chinese owners sympathetic to or otherwise compelled to submit to a political leader’s will.
Is it possible for a potentially vile political dictator to, hypothetically, choose to deliberately manipulate the pharmaceutical supply of a country adverse to them as a military/political strategy? Does that represent a National Security threat?
Unscientific Extrapolation of Quality Control Findings:
To assist in the illusion of quality control security to consumers, in July of 2022, the FDA published a farcical, obviously staged, internal interview with its OPQ Director of Surveillance in charge of quality control testing, Dr. Jennifer Maguire.
In her interview, Dr. Maguire categorically declared that “a very low percentage of drugs did not meet quality standards.” In stating so, Dr. Maguire referenced a study that her FDA supervisor and OPQ director, Dr. Michael Kopcha (a pharmacologist and pharmacist) had co-authored. That study had tested a mere 252 products, containing only 17 different active ingredients…out of the 150,000 total products that the OPQ is tasked with policing for quality.
Even if one knows nothing about pharmaceutical quality testing, ponder the following: in what (if any) quality control scenario is it suitable to test a total of 252 non-unique products out of 150,000 (that is: ~0.001%, or one one-thousandth of one percent) of highly variable, highly complex, and highly regulated product, then extrapolate ~0.001% of those findings to 150,000 products, which in turn are made at hundreds of different locations around the world with varying staff, education, training, oversight, equipment, and records of violations?
It’s easily considered scientifically unsound methodology, yet Dr. Maguire delighted about how her department had conducted “the largest known sampling study of its kind.” That means the FDA seems to have permitted this kind of extrapolation for decades.
Even worse: that publication’s methodology seems even more irresponsible when considering that of those 217 generic drugs in that study used to justify her assertion in 2022, only ~5% were from Asia and only 36% were from India, despite those countries making very nearly 100% of non-narcotic generic drugs and/or at least one of their “building-block” components that fill American hospitals and drugstores.
Is there any technically complex manufacturing model anywhere that tests quality control in a manner so trusting – let alone that of pharmaceuticals – upon which America’s pharmacists and physicians depend on to treat elderly, infirm, and hospitalized patients’ critical and/or fragile medical conditions?
The answer is obviously no, but based on the OPQ’s publication plus yearly website inspection records released to the public, it appears that is exactly the methodology that the FDA leadership is implementing.
Barring my misinterpretation of FDA publications and shared testing data, it might be the worst example of confirmation bias I have seen in my scientific career.
Obviously the FDA’s OPQ surveillance website claim of “We at CDER are committed to protecting patients and consumers from unsafe, ineffective or poor-quality drugs” is widely discordant from their real-world practice of seemingly independently collecting and testing only 0.001% of the products they regulate.
The question is: Who at the taxpayer-funded FDA concluded that this represents a scientifically sanctified testing methodology?
Scant Drug Testing by America’s FDA Despite Poor Quality:
The manifestations of OPQ’s nearly non-existent drug quality surveillance policies have been yielding negative patient outcomes for a long time now.
A December 2023 publication detailed that the FDA recalls related to drug manufacturing quality more than doubled in the US from 2018 to 2022. Specifically, according to STAT news (an ancillary publication of the Boston Globe) in 2018, there were only 22 product recalls, but in 2022, there were 310 – a 14x increase. That number would have almost certainly been higher, with a broader inclusion plus independent testing of Chinese and Indian drugs.
In the cases listed, the FDA only recalled those products after they had affected consumers. Most, if not every single FDA recall could have potentially been avoided, had the FDA been directly and more comprehensively testing products prior to them reaching distributors, hospitals, or pharmacies.
Deaths and Serious Injuries Caused by Lack of Independent Collection and Testing of Overseas Pharmaceuticals by the FDA:
The failure of the FDA to independently confirm product quality lies directly at the auspice of FDA leadership. The FDA’s duty to guarantee drug purity dates back to the original reason it was founded back in 1906. Back then, the Pure Food Drug and Cosmetic Act was codified into law by Congress, mostly to assure that product ingredients were listed, and listed accurately. Assuring ingredients is still a critical part of the FDA’s mission, and someone needs to remind them of the same.
When the FDA ignores its duties to assure quality control across a wide breadth of products, extremely serious and/or deadly consequences occur. The following list of examples is in no way complete and includes:
- A November 2023 report detailed how artificial tears made in India, widely sold at Walmart, CVS, and Target (as in-house brands) had to be recalled from pharmacy shelves after being found to be produced in an unsanitary facility in India where manufacturers forged test results they submitted to regulators.
- Separately, in 2022, tainted artificial tears imported from India caused four Americans to die and 18 others to lose their vision. In some cases, the infections were so severe that patients had to have their eyeballs surgically removed from their sockets.
- On February 5th, 2024 yet another urgent (Class 1) recall was issued for 27 widely available eye drops. A Class I designation is the FDA’s most serious recall category and warns use of the recalled products could cause “…serious adverse health consequences or death.” The eye drops were produced overseas and distributed widely to consumers by just about every chain pharmacy in the United States.
- In June 2024 Scores of CVS and Cardinal Health-branded products were recalled due to selling infected eye drops. In what was supposed to be a sterile manufacturing facility (think: hospital surgical operating room) an inspector witnessed workers walking around barefoot and “brushing [their] hair.”
- In another June 2024 inspection, nasal sprays intended for infants shared equipment used to make pesticides.
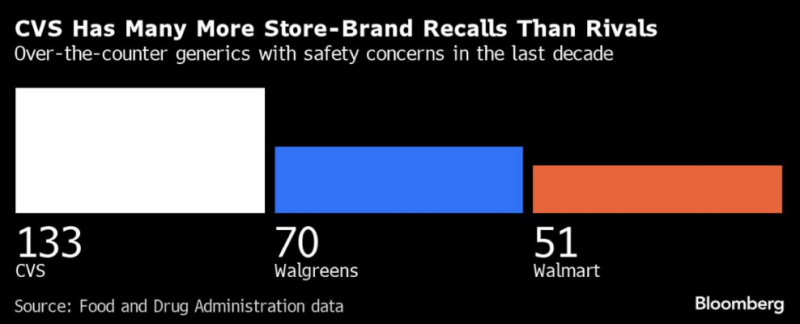
- In the last 10 years, CVS has recalled 133 over-the-counter generic drugs due to safety concerns. CVS has outsourced production of generic drugs to at least 15 manufacturers that have been cited for problems, according to Bloomberg. Other retail pharmacies that sell house-branded products faired only somewhat better:
- In 2019, an inspection by the Food and Drug Administration found that medicine was produced in a facility in India where workers walked around barefoot, paint was peeling off the walls, and bacteria was turning up in samples. The FDA response was to simply issue a warning to consumers not to take the eye drops back in late October 2019, but they remained on store shelves at least two weeks after the fact. Patients who had taken eye drops made out of that factory experienced vision loss. Obviously, these eye drops should have never been on the shelves at all, let alone two weeks following an FDA recall!
- In June of 2023 Gujarat, Ahmedabad, India-based Intas Pharma, a key supplier of chemotherapeutic drugs to the US such as cisplatin and carboplatin, was found to have gross manufacturing and data integrity issues resulting in numerous FDA warning letters. As a result of Intas producing poor quality products, patients already suffering from gynecological, head, neck, and bladder malignancies went untreated and/or may have been given adultured chemotherapeutic products. No other supplier of these products were available since Intas had undercut its manufacturing competitors in the US and elsewhere, putatively by cutting corners to save money. The FDA placed Intas Pharma on an “import alert” which entails sending a widespread communication to providers, typically in the outdated modality of sending a letter via US mail. While the FDA “import alert” stops new products coming into the US, it does not seize or “recall” existing products which are already present in hospitals, pharmacies, and in physician offices.
- In July of 2023, dextromethorphan-based cough syrups made by Maiden Pharmaceuticals Ltd, an older, established drug company in Pitampura, New Delhi, India were found to be produced with toxic industrial solvents including as diethylene glycol and ethylene glycol as contaminants. Maiden’s cough syrups were distributed over 18 months in 10 countries, resulting in the deaths of at least 66 children on top of the untold hundreds of children suffering permanent kidney damage.
According to reports: the facility which produced the cough syrup:
“…repeatedly failed quality tests in the years leading up to the outbreak. One substandard-drugs case dragged through India’s sclerotic courts for nine years before being dismissed on a technicality. Another [case] took 12 years and resulted in a fine of 1,000 rupees, or about $15 at the time.”
Generally speaking, 2024 pharmaceutical quality of products originating in India continue to persist and have been aptly described by pharmaceutical quality experts as “sad” and by pundits as an “unending saga of quality lapses.” Those lapses are directly due to poor manufacturing controls, compounded on top of the FDA’s profound lack of oversight in the form of live, in-person inspections and analytical testing of end products as they are imported into the United States. Is it enough to wake the FDA from its quality control duty slumber? Will the FDA have any reaction other than to ask for “mailed-in” sampling?
Are the above cases – including those that resulted in death/blindness in 2024 – into what Dr. Maguire glibly dismisses as “low percentage” of drugs which “did not meet quality control standards?” Indeed, had the FDA been using its prior six-plus billion dollar yearly budget – half of which is discretionary spending – to more comprehensively inspect and/or test this and other eye drops before they reached pharmacies, those deaths and blindness cases may have been prevented.
Both manufacturers and the FDA leadership team’s behavior was predicted about 25 years ago through the great Thomas Sowell who stated:
It is hard to imagine a more stupid or more dangerous way of making decisions than by putting those decisions in the hands of people who pay no price for being wrong.
Ironically enough, Sowell was then, too, referring to drug safety with his quote.
In fact, the only meaningful price one paid for being wrong when it came to poor manufacturing I could find in the recent past was here in the United States.
It involved New England Compounding Centers pharmacists Glenn Chin and pharmacy supervisor Barry Gadden, who are each facing around a decade in prison for manslaughter after they produced contaminated steroids at their independent pharmacy, in yet another example of targeting a small business while Big Pharma overseas manufacturers are not prosecuted for potentially equal or even worse crimes.
What (if anything) will ever happen to Chinese and Indian manufacturers – or FDA employees – who fail to perform their oversight jobs?
Why aren’t they being held accountable for serious injuries and/or deaths?
…What about Outside, Third-Party Drug Quality Testing?
Since the FDA isn’t conducting independent collecting and testing, what about going to independent laboratories?
While there are outside laboratories that advertise pharmaceutical testing services, one such company, Valisure, while purporting to protect Americans from poor quality products, actually seemed to be more about “shaking down” manufacturers for cash in place of ignoring protecting Americans, according to the Wall Street Journal. Still, that has not stopped Valisure from proposing to test for “impurities” in America’s drugs – but impurities are not all-inclusive and are only part of the story because it does zero to address quantitative (milligram strength) excursions or biological contamination.
Despite Valisure’s name (a seeming amalgamation of “validation” and “assurance”) it provides neither. Its so-called laser-testing methodology was developed outside of Valisure. Plus Valisure’s laser-testing methodologies are highly exothermic, which would degrade and/or create toxic byproducts. Heat is well-known to alter the composition of many chemicals, including drugs. Valisure’s entire business model had been scolded by judges, legal scholars, and scientific experts as “junk science” as well as by the Wall Street Journal’s editorial board and by an independent analytical chemist.
As if that wasn’t enough, Valisure was also accused of committing a multitude of regulatory violations by the FDA’s legal/compliance department, having been accused of making false scientific claims in court.
Bottom line: Americans are more-or-less fully dependent on the FDA and/or their specifically certified laboratories to assure America’s pharmaceutical quality. In the FDA’s absence, outside, low-rent, self-labeled “quality assurance” services (including Valisure) will take advantage of substantial FDA gaps in testing and transparency to promote their testing services to ignorant clientele, which alarmingly includes the Pentagon.
Employing GMP-certified pharmaceutical release testing (a term of art) is the only way to appropriately test a pharmaceutical product for all qualitative and quantitative excursions. It’s something that the FDA is obligated to do as part of its public health mission, and additionally has both the personnel on top of discretionary spending to do so. Valisure’s website gives no indication that it has even attempted to become certified as an official “pharmaceutical release testing” facility.
…What about Lay Consumers Trying to Conduct Their Own Quality Investigations Via the FDA’s Inspection Reports?
Consumers – even those with expertise in analytical chemistry and quality control – attempting to select preferred brands based on the country of origin by reading FDA inspection reports would have great difficulty in interpreting findings.
Alternatively, attempting to preferentially select a US or European manufacturer with better accountability and/or safety records wouldn’t work either, because even if one was available, pharmacy insurance plans typically only reimburse for the cheapest manufacturer of any generic product or therapeutic class. That likely translates to something made at a Chinese or Indian manufacturing facility.
Even if pharmacy insurance was not an issue, attempting to dig into FDA records and investigate manufacturers would have roadblocks such as the following example:
A September 2023 violation issued by the FDA only gave American consumers an excruciatingly over-redacted inspection report from the Sichuan Deebio Pharmaceutical plant in China. During that particular FDA inspection, the plant’s quality control chief appeared to lie (or as inspectors politely described it, gave “misleading information”) about critical test results and recordkeeping.
Sichuan Deebio’s quality control supervisor reversed her official statements regarding quality control records and recordkeeping made to FDA inspectors, (more than once) eventually reverting back to her original statement. In the end, she admitted to “not telling the truth about recording the results on respective data worksheets,” further admitting that an essential record/worksheet never even existed. To make matters worse, investigators subsequently asked the quality leader how she kept track of those highly-specific test results, to which she replied that they were “in her mind.”
The FDA’s publicly available report detailing other particulars of that inspection is mostly unreadable following seemingly hyperactive “(b)(4)” redactions by the FDA. Of note, “(b)(4)” redactions are considered by the FDA to be “trade secret” even though the inspections are directly related to public health and taxpayer-funded.
Furthermore, there exists a lengthy history of FDA documents, letters, and reports which after FDA redactions read more like top-secret Pentagon or CIA reports or 1950s children’s “Mad Libs” word games rather than critical HHS/public health documents which exist to inform the public of important safety problems.
To that end, here are some excerpts from the FOIA’d versions of the Sichuan Deebio plant FDA inspection report. Following redactions, it’s generally impossible to make any sense of what particular impurities or degree of impurities were uncovered by FDA officials:
Even the batch numbers affected were retracted, preventing consumers from doing their own check of which particular drugs/lots were affected.


Further looking around on the internet for more information shows the Sichuan Deebio’s external English-version of its website to be uncomically grammatically incoherent, and/or appearing to have photographs of its facility which are stock and/or fake, and/or obviously computer-generated and/or logistically unrealistic. There are clearly many warnings, further questioning the credibility and truthfulness of just about anything that Sichuan Deebio represents.


Of note, Sichuan Deebio products are mainly designed and manufactured for Western (United States) markets.
The worst part of it is: with its overt disregard for recordkeeping, quality, or the FDA, it doesn’t appear that Sichuan Deebio was blocked by the FDA from continuing to sell its downstream inventory already shipped in US hospitals and pharmacies.
No Shortage of FDA Personnel or Funding for Testing, Unacceptable Methodology…Yet, Employees Get Taxpayer-Funded Raises:
The FDA’s lack of independent, comprehensive pharmaceutical testing is not due to a lack of FDA personnel, funding or other resources. It’s due to senior leadership’s seeming indifference of not being accountable to anyone for how they spend their funding.
Most recently figures show that the FDA’s 2024 budget swelled to $7.2 billion, about half of which is fully discretionary.
In lieu of implementing widespread prospective testing to protect Americans, the FDA has instead given its nearly 20,000 employees some of the highest salaries of any government agency – and that’s on top of its:
- 3.1% pay raise in 2020,
- plus a 2.7% pay raise in 2022,
- plus a 4.6% percent pay raise in 2023
- plus a 5.2% pay raise in 2024.
Back in 2022, the FDA was already 108th in the entire US for overall highest-paying employers; it’s a foregone conclusion that FDA employees have further improved on that figure as of today.
Summary:
Trusting for-profit, overseas manufacturers to police themselves on quality control via self-selected “mailed-in” sampling doesn’t work because it conflicts with a manufacturer’s fundamental incentive for profit. Patients filling prescriptions hoping to treat a critical medical condition could instead be adversely affected with toxic, incorrect strength, or otherwise impure products stealthily and perniciously making them even sicker.
Pharmacists and physicians have enough complex patient care responsibilities and shouldn’t have to additionally consider the possibility of toxic/poor quality drugs as an additional potential complicating factor.
The collaboration of President Donald Trump and Robert F. Kennedy, Jr. to Make America Healthy Again (MAHA) is a massive and critically necessary endeavor. Part of that must also implement immediate and far more stringent oversight of Chinese and Indian pharmaceuticals.
The two best ways to advance drug quality exist at the intersection of independent collection and verification by the FDA before they get to patients along with Donald Trump’s continued advocacy to return pharmaceutical manufacturing back to the United States where manufacturers can be better monitored and held accountable if errors take place. In addition to reshoring high-tech jobs, it would allow the FDA routine and immediate access to manufacturing plants for unannounced inspections. In fact, that endeavor had already begun with Trump’s initiatives via the Phlow Corporation to manufacture precursor “building blocks” for pharmaceutical manufacturing.
With decades-plus-long, legendary track records of poor quality and fraud in the examples listed here, how could the FDA not test everything coming out of China or India without independent collection and conclusive and transparent quality verification? It’s not only irresponsible to patients, pharmacists, and physicians who depend on the FDA to verify drug quality to improve health – a lack of quality-verified pharmaceuticals represents a National Security threat to all Americans as well.
Obviously, independent and prospective selection and testing of all drugs for quality assurance by batch/lots in real time at ports of entry, distribution hubs, or wholesalers – before they are sent off to hospitals or pharmacies – on top of regular, live manufacturing facility inspections is the most sensible strategy.
Until the FDA takes a sincere, boots-on-the-ground and proactive methodology to prevent bad products from hitting pharmacy shelves in the first place, Americans can expect unending, after-the-fact recalls, permanent injuries, and deaths directly related to the FDA’s existing ad-hoc, remote, minimal and methodologically unsanctified testing and supervision.
As for today – and to answer the question posed near the beginning of the article – who independently collects and verifies drug quality from China and India?
Answer: It’s NOT our FDA.
DISCLAIMER: This article is not medical advice. Do NOT start or discontinue ANY drug without first discussing it with a pharmacist or physician you know and trust.
Join the conversation:
Published under a Creative Commons Attribution 4.0 International License
For reprints, please set the canonical link back to the original Brownstone Institute Article and Author.








